The following paper was originally published in Journal of Clinical Oncology in 2018. Researchers compare clinical, radiologic, and histomolecular characteristics between short-term survivors and long-term survivors of DIPG using data compiled from the International DIPG /DMG Registry and their 1,100+ patient records collected. The DIPG/DMG Registry is fully funded by the DIPG Collaborative and The Cure Starts Now. A 5 country alliance of over 100 hospitals and over 1,100 DIPG/DMG patients, it is the definitive linked database on DIPG and DMG that is already changing how we win against cancer.
Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries
Originally published in JOURNAL OF CLINICAL ONCOLOGY
Lindsey M. Hoffman, Sophie E.M. Veldhuijzen van Zanten, Niclas Colditz, Joshua Baugh, Brooklyn Chaney, Marion Hoffmann, Adam Lane, Christine Fuller, Lili Miles, Cynthia Hawkins, Ute Bartels, Eric Bouffet, Stewart Goldman, Sarah Leary, Nicholas K. Foreman, Roger Packer, Katherine E. Warren, Alberto Broniscer, Mark W. Kieran, Jane Minturn, Melanie Comito, Emmett Broxson, Chie-Schin Shih, Soumen Khatua, Murali Chintagumpala, Anne Sophie Carret, Nancy Yanez Escorza, Timothy Hassall, David S. Ziegler, Nicholas Gottardo, Hetal Dholaria, Renee Doughman, Martin Benesch, Rachid Drissi, Javad Nazarian, Nada Jabado, Nathalie Boddaert, Pascale Varlet, G´eraldine Giraud, David Castel, Stephanie Puget, Chris Jones, Esther Hulleman, Piergiorgio Modena, Marzia Giagnacovo, Manila Antonelli, Torsten Pietsch, Gerrit H. Gielen, David T.W. Jones, Dominik Sturm, Stefan M. Pfister, Nicolas U. Gerber, Michael A. Grotzer, Elke Pfaff, Andr´e O. von Bueren, Darren Hargrave, Guirish A. Solanki, Filip Jadrijevic Cvrlje, Gertjan J.L. Kaspers, William P. Vandertop, Jacques Grill, Simon Bailey, Veronica Biassoni, Maura Massimino, Rapha¨el Calmon, Esther Sanchez, Brigitte Bison, Monika Warmuth-Metz, James Leach, Blaise Jones, Dannis G. van Vuurden, Christof M. Kramm, and Maryam Fouladi
Abstract
Purpose
Diffuse intrinsic pontine glioma (DIPG) is a brainstem malignancy with a median survival of 1 year. The International and European Society for Pediatric Oncology DIPG Registries collaborated to compare clinical, radiologic, and histomolecular characteristics between short-term survivors (STSs) and long-term survivors (LTSs).
Materials and Methods
Data abstracted from registry databases included patients from North America, Australia, Germany, Austria, Switzerland, the Netherlands, Italy, France, the United Kingdom, and Croatia.
Results
Among 1,130 pediatric and young adults with radiographically confirmed DIPG, 122 (11%) were excluded. Of the 1,008 remaining patients, 101 (10%) were LTSs (survival $ 2 years). Median survival time was 11 months (interquartile range, 7.5 to 16 months), and 1-, 2-, 3-, 4-, and 5-year survival rates were 42.3%(95%CI, 38.1%to 44.1%), 9.6%(95% CI, 7.8% to 11.3%), 4.3% (95% CI, 3.2% to 5.8%), 3.2% (95% CI, 2.4%to 4.6%), and 2.2% (95%CI, 1.4%to 3.4%), respectively. LTSs, compared with STSs, more commonly presented at age 3 or 10 years (11%v 3% and 33% v 23%, respectively; P = .001) and with longer symptom duration (P , .001). STSs, compared with LTSs, more commonly presented with cranial nerve palsy (83%v 73%, respectively; P = .008), ring enhancement (38% v23%, respectively; P = .007), necrosis (42%v 26%, respectively; P = .009), and extrapontine extension (92% v 86%, respectively; P = .04). LTSs more commonly received systemic therapy at diagnosis (88% v 75% for STSs; P = .005). Biopsies and autopsies were performed in 299 patients (30%) and 77 patients (10%), respectively; 181 tumors (48%) were molecularly characterized. LTSs were more likely to harbor a HIST1H3B mutation (odds ratio, 1.28; 95% CI, 1.1 to 1.5; P = .002).
Conclusion
We report clinical, radiologic, and molecular factors that correlate with survival in children and young adults with DIPG, which are important for risk stratification in future clinical trials.
Introduction
Diffuse intrinsic pontine glioma (DIPG) is a malignant brainstem tumor of childhood for which median survival is, <1 year.1 Longterm survival, historically defined as overall survival (OS) >2 years, is reported in <10% of patients.1 Characteristics associated with longer survival include younger age, longer symptom latency, and absent ring enhancement on diagnostic magnetic resonance imaging.1,2 Up to 90% of DIPGs harbor a pathognomonic point mutation in H3F3A (65% of tumors) or HIST1H3B (25% of tumors); the latter seems to confer longer survival. Ten percent of patients have a histone 3 wild-type tumor.3
Involved-field radiation therapy (RT) remains standard of care but confers only a 3- to 4-month survival advantage. Benefit from neoadjuvant4 or adjuvant2,5 chemotherapy has not been consistently confirmed in prospective trials.
The rarity and inconsistent classification of DIPG, an imaging based diagnosis, have long hampered cross-cohort comparisons. The primary aim of this multinational collaboration between the International DIPG Registry (IDIPGR) and European Society for Pediatric Oncology DIPG Registry (SIOPE-DIPGR)6,7 was to define clinical, radiologic, histologic, and molecular factors associated with short- and long-term survival in the largest cohort of centrally reviewed DIPGs to date.
Materials and Methods
Study Population
The study was approved by the institutional review board at Cincinnati Children’s Hospital Medical Center and included 1,130 patients with radiographically confirmed DIPG diagnosed from 1990 to 2015. IDIPGR patients (n = 409) were age 0 to 27 years from the United States, Canada, and Australia. SIOPE-DIPGR patients (n = 721) were age 0 to 21 years from the Netherlands, Germany, Austria, Switzerland, Italy, France, the United Kingdom, and Croatia. Patients were referred to the registries as previously described.6,7 Exclusion criteria are listed in Figure 1. No patients with neurofibromatosis type 1 were included.
Clinical Variables
Clinical data were abstracted (J.B., B.C., S.E.M.V.v.Z., and N.C.) using standardized case report forms. Cerebellar signs included dysmetria, ataxia, dysarthria, or nystagmus. Pyramidal tract signs included mono-, hemi-, or quadriparesis; hyperreflexia; or positive Babinski sign. Because over survival (OS), defined as the time from diagnosis to death or last follow-up, is regarded as the most reliable outcome variable for DIPG, progression-free survival (PFS) was not reported. Short-term survivors (STSs), long-term survivors (LTSs), and very long–term survivors (VLTSs) had OS times of, <24, ≥24, and ≥60 months, respectively. Two LTSs (patients DIPG-0016 and DIPG-0081) lost to follow-up at our data cutoff (January 1, 2017) were included in primary statistical analyses.
Radiologic Variables
Anonymized diagnostic magnetic resonance imaging was centrally reviewed (M.W., B.B., E.S., R.C., J.L., and B.J.) and classified as typical or unlikely DIPG; the latter were excluded. Typical DIPGs arose from and diffusely involved ≥50% of the pons. Exclusionary features included
focally exophytic morphology, marked diffusion restriction, or secondary brainstem involvement by a tumor centered elsewhere in the brain or spine. Diagnostic imaging from all LTSs and 10% of STSs was cross-validated by a neuroradiologist from the other registry. Metastatic disease, defined as noncontiguous tumor in the brain or spine, was reported by individual sites but not centrally reviewed.
Histopathologic and Molecular Variables
Histology was defined according to 2007 WHO criteria; based on availability of tissue in the registries, 61 tumor specimens were centrally reviewed (C.F. and C.H.). Databases were queried for common genomic alterations in DIPG. Histone mutations were assessed by Sanger sequencing, whole-exome sequencing, or whole-genome sequencing, polymerase chain reaction, or immunohistochemistry to detect H3K27M-mutant protein or H3K27 trimethylation (H3K27me3). Mutations in H3F3A (H3.3 K27M) or HIST1H3B (H3.1 K27M) were considered mutually exclusive even if both were not evaluated.
Statistical Analyses
Patient characteristics were summarized using medians and ranges or frequencies and percentages. Univariable analyses were performed using the Fisher’s exact test or Wilcoxon rank sum test. Multivariable logistic regression was performed on variables with <15% missing data and univariable P <.1; however, transverse tumor dimension was excluded as a result of high correlation with craniocaudal dimension. For subgroup analyses, multivariable logistic regression models were used to determine subgroup significance and adjusted for confounding factors. Survival was estimated using the Kaplan-Meier method. Statistical evaluation was performed using R (Version 3.1.3). P <.05 was considered significant.
Results
Survival
A total of 1,008 patients met inclusion criteria (IDIPGR, n = 374; SIOPE-DIPGR, n = 634). Median survival time was 11 months (interquartile range, 7.5 to 16 months), and 1-, 2-, 3-, 4-, and 5-year OS rates were 42.3% (95% CI, 38.1% to 44.1%), 9.6% (95% CI, 7.8% to 11.3%), 4.3% (95% CI, 3.2% to 5.8%), 3.2% (95% CI, 2.4% to 4.6%), and 2.2% (95% CI, 1.4% to 3.4%), respectively. Characteristics of 101 LTSs (10%) and 16 VLTSs (1.6%) are shown in Figure 2 and Appendix Figure A1 (online only), respectively. Kaplan- Meier survival analyses for age, symptom duration, systemic therapy, histology, and molecular status are shown in Figure 3.
Clinical Presentation Median age was 6.8 years (range, 0 to 26.8 years); 4% of patients were age <3 years at diagnosis. Of patients with available data, 755 (82%) of 917, 468 (51%) of 915, and 567 (62%) of 920 patients presented with one or more cranial nerve (CN) palsy, pyramidal tract, or cerebellar sign, respectively. On univariable analysis (Table 1), LTSs were more likely to be age <3 years (28% v 3% of STSs) or >10 years (33% v 23% of STSs; P = .001) and had longer symptom duration at diagnosis. LTSs were less likely to present with CN palsy (72% v 83% of STSs; P = .008). Multivariable analyses (Table 2) confirmed association of age and symptom duration with long-term survival but failed to associate CN palsy with short-term survival.
Therapy
Thirty-eight patients (3%) who did not receive therapy at diagnosis (Appendix Fig A2A, online only) were excluded. Untreated patients were more often <3 years old at diagnosis. Eleven patients underwent biopsy or autopsy. At progression, one patient received chemotherapy; no patients received RT. Median OS of untreated patients was 1 month (range, 0 to 135 months). Two patients were LTSs (both infants), including one who was alive 135 months after diagnosis (Appendix Fig A2B, online only).
The status of RT and systemic therapy was known for 968 patients; 721 patients (74%) received both RT and systemic therapy, 231 patients (24%) received RT alone, and 16 patients (2%) received systemic therapy alone. In univariable and multivariable analyses, LTSs more commonly received systemic therapy at diagnosis (88% v 75% for STSs; P = .005; odds ratio [OR], 3; 95% CI, 1.46 to 7.3; P = .01). Systemic therapy type was known for 702 patients (70%); 350 patients (50%) received cytotoxic therapy only, 193 patients (27%) received targeted therapy only, and 159 patients (23%) received both cytotoxic and targeted. On univariable analysis, type of targeted therapy yielded no survival difference (Table 1). However, multivariable logistic regression adjusted for age and symptom duration demonstrated greater odds of long-term survival with use of an epidermal growth factor receptor (EGFR) inhibitor (OR, 2.32; 95%CI, 1.1 to 4.82; P = .03) or bevacizumab (OR, 2.67; 95%CI, 1.09 to 6.55; P = .03), an anti–vascular endothelial growth factor (VEGF) antibody, at diagnosis (Table 2). Seventy-two patients (7%) underwent reirradiation at first or subsequent progression (as reported by individual sites). The rate of first progression recorded within 1 year of diagnosis was significantly lower in patients who underwent reirradiation compared with patients who did not (74% v 88%, respectively; P = .007).
Imaging
Table 1 lists diagnostic imaging characteristics. STSs demonstrated larger craniocaudal tumor dimension (43 v 40 mm for LTSs; P = .04) and higher rates of extrapontine extension (92% v 85%for LTSs; P = .04), tumor necrosis (45% v 26% for LTSs; P = .009), and ring enhancement (38% v 23% for LTSs; P = .007). Metastatic disease at diagnosis was reported in 18 STSs (2%) and no LTSs.
Histology and Molecular Characteristics
More SIOPE-DIPGR patients (39%) than IDIPGR patients (14%) underwent biopsy, and more IDIPGR patients (16%) than SIOPE-DIPGR patients (4%) underwent autopsy (Appendix Table A1, online only). LTSs from both registries were more often biopsied than STSs (38% v 28%, respectively; P = .04). Histology and WHO grade were known for 288 biopsy and 76 autopsy samples. WHO grade did not influence survival. Biopsy specimens included glioblastoma multiforme (GBM; n = 80), anaplastic astrocytoma (n = 76), anaplastic oligodendroglioma (n = 10), diffuse astrocytoma (n = 37), fibrillary astrocytoma (n = 4), oligodendroglioma (n = 2), low-grade astrocytoma (n = 8), and unknown (n = 71). Histology of autopsy tissue included GBM (n = 48), anaplastic astrocytoma (n = 12), diffuse astrocytoma (n = 3), and unknown (n = 13).
Of 376 patients from whom tissue was obtained, genomic data were available for 181 (48%) of patients (18% of the entire cohort; Data Supplement), including 21 LTSs (Fig 4). Global molecular assessment was undertaken for 44 patients (whole-genome sequencing, n = 16; whole-exome sequencing, n = 25; 450k methylation array, n = 3), whereas 98 patients underwent limited genomic sequencing (Sanger, n = 80; other targeted platform, n = 18), and 36 patients underwent immunohistochemistry alone. H3.1 K27M was associated with longer median OS (15 months) and long-term survival in multivariable analysis (OR, 1.28; 95% CI, 1.1 to 1.5; P = .002). In contrast, H3.3 K27M was associated with short-term survival (OR, 0.88; 95% CI, 0.78 to 0.99; P = .04; median survival, 10.4 months). Patients with H3 wild-type tumors (n = 26) had a median OS of 10.5 months. WHO grade did not correlate with histone mutation status. TP53 and ACVR1 mutations were not associated with survival. Of the 50 patients age 10 years at diagnosis, who as a group demonstrated higher likelihood of long-term survival, 38 (78%) harbored H3.3 K27M, nine (18%) were H3 wild-type, and only three (6%) had H3.1 K27M.
Discussion
This study confirms the relevance of some previously reported survival-associated factors in patients with DIPG and offers unique insight into 101 LTSs (including 16 VLTSs). Median survival for all 1,008 patients was 11 months.1,5 Median survival times of LTSs and VLTSs were 33 months (range, 24 to 156 months) and 78 months (range, 60 to 156 months), respectively. Of 16 surviving patients, two were lost to follow-up but were LTSs at the time of last contact (patients DIPG-0016 and DIPG-0081; OS, 33 and 36 months). The 2-year OS rate of 9.6% in this study was consistent with large retrospective studies2,5 that reported 9.2% and 9% 2-year OS rates in 153 and 316 patients with DIPG, respectively. The 1-year OS rate in our study (42.3%) is comparable to that reported by Hassan et al9 in a meta-analysis of 2,336 pediatric patients with high-grade brainstem glioma (41%); however, the 2- and 3-year OS rates of 15.3% (95% CI, 12% to 20%) and 7.3% (95% CI, 5.2% to 10%) in their study were higher than those in our study (9.6% and 4.3%, respectively), likely reflecting the heterogeneity of their cohort, some whom may not have true DIPGs.
Previously, 43 VLTSs had been reported in the literature.1,10-15 In Appendix Figure A1, we compare the characteristics of 22 previously published VLTSs to our 16 VLTSs, including eight (0.02% of the total cohort) who are alive with a median follow-up time of 6.5 years (range, 5 to 13 years). Our 5-year OS rate of 2.3% is comparable to the rate of 2.6% reported by Jackson et al1 in 191 patients with DIPG; however, two of their five VLTSs would have been excluded from our study for atypical magnetic resonance imaging features. Freeman et al12 reported nine VLTSs (6.9%) among 130 patients with DIPG treated with hyperfractionated RT (Pediatric Oncology Group 8495 trial), although only four of these patients (3%) would have met inclusion criteria in our study.
Age <3 or >10 years, longer symptom latency, lack of CN palsy, and systemic therapy at diagnosis were predictors of longterm survival. Of 41 patients age 3 years at diagnosis, 36 received first-line RT with or without systemic therapy and five received systemic therapy alone. Although median OS for children age 3 years (11 months) was the same as the entire cohort, a greater proportion was LTSs or VLTSs. Other studies have reported similar findings.1,2,5,16 Broniscer et al17 described 10 DIPG patients age 3 years who received RT with or without chemotherapy (n = 8) or chemotherapy only (n = 2) at diagnosis (n = 6) or progression (n = 4).
Five patients (50%) were LTSs, including one treated without RT. Wagner et al5 similarly reported higher median survival in 13.
Patients age >10 years at diagnosis had longer median OS (13 months) and were more likely to be LTSs. Bailey et al19 similarly reported five LTSs (all >9 years old) among 43 patients with radiographically confirmed DIPG. In contrast, Veldhuijzen van Zanten et al16 reported no difference in OS between patients age 9 to 18 years versus younger patients. Although pathogenic mechanisms, such as low-grade histology or IDH mutation may influence survival in older patients, 78% of patients > 10 years old in our study harbored the poor prognostic H3.3 K27M mutation. Clinical and molecular characteristics for patients age > 18 years (n = 13) were also similar to their younger counterparts (Appendix Fig A3, online only).
Consistent with prior reports,1,2 the presence of symptoms for >24 weeks at diagnosis was strongly associated with longer survival in univariable and multivariable analyses. CN palsy at diagnosis predicted shorter survival in univariable but not multivariable analysis. Previous studies reporting association of CN palsy with shorter survival included all brainstem tumors, not just DIPG, and/or diagnosis based on computed tomography scan, making comparison difficult.20
Neoadjuvant or adjuvant systemic therapy correlated with long-term survival in both univariable and multivariable analyses. This finding differs from the long-standing view that systemic therapy provides no survival benefit for DIPG, a principle largely based on small, nonrandomized clinical trials. Effective crosscomparison of therapeutic studies for DIPG has been hindered by wide variation in inclusion criteria, as demonstrated in studies by Hargrave et al21 and Jansen et al22 in which only six of 29 DIPG specific therapeutic trials between 1984 and 2012 had comparable eligibility. In a randomized trial, Wagner et al5 reported better median OS in patients with DIPG treated with adjuvant chemotherapy after RT (11.3 months) compared with patients treated with RT alone (9.5 months; P = .03). Similarly, others have reported superior median OS with use of adjuvant or neoadjuvant chemotherapy.4
Multivariable logistic regression demonstrated higher odds of long-term survival with use of EGFR inhibitors (eg, gefitinib, erlotinib, nimotuzumab, rindopepimut, cetuximab) or bevacizumab at diagnosis. A phase II study of gefitinib with RT in newly diagnosed patients with DIPG noted 2-year OS of 19.6%with PFS.36 months in three patients.23 In a biopsy-mandated phase I study of erlotinib with RT, EGFR overexpression trended toward longer PFS (10.1 months v 6.3 months in patients without EGFR overexpression; P = .058) but not OS.24 Despite only modest activity of nimotuzumab in progressive DIPG, two patients lived for 663 and 481 days from the start of therapy.25
Despite efficacy in adult GBM, bevacizumab has shown little activity in pediatric trials for newly diagnosed26 or progressive DIPG27 (median PFS, 2.3 months). However, in a phase I trial of vandetanib, a selective vascular endothelial growth factor receptor receptor 2 (VEGFR2) and EGFR inhibitor, in newly diagnosed DIPG, Broniscer et al28 reported 2-year OS of 21.4%, and higher levels of plasma VEGF were associated with longer PFS (P = .02). Although numbers were too small to assess patient outcomes based on genomically matched targeted therapy, our findings support prospective assessment of biopsy tissue to define potential therapeutic targets, as recently undertaken in two multi-institution, multinational trials (ClinicalTrials.gov identifiers: NCT01182350 andNCT02233049).
Janssens et al29 reported improved OS in 31 children with DIPG who received reirradiation at first progression (13.7 months) compared with a matched control cohort (10.3 months) despite similar PFS (8.2 v 7.7 months, respectively). Progression was not defined or centrally reviewed in our study; however, we noted that the proportion of patients with recorded progression within 1 year of diagnosis was significantly lower among patients who underwent reirradiation compared with those who did not, suggesting potential clinician bias to recommend reirradiation to patients with a more indolent disease course or potentially greater sensitivity to initial RT in patients who ultimately received reirradiation. As postulated by others,30 increased RT sensitivity may be a manifestation of distinct biology. We did not report reirradiation-based outcomes given limitations conferred by analysis of registry data; more robust analysis of the effect of reirradiation in patients with DIPG would be best assessed prospectively in the context of a clinical trial.
On the basis of the radiographic definition of DIPG by Barkovich et al,31 patients with <50% pontine involvement (n = 5) were excluded. Similar to a prior report,5 these patients had better median OS (20 months), and two patients were LTSs. Greater craniocaudal tumor dimension and extrapontine extension were associated with shorter survival; the former finding contrasts with a report by Poussaint et al,32 in which larger tumor at diagnosis was associated with longer survival.
As previously described,32 tumor necrosis and ring enhancement were associated with short-term survival in univariable analysis. Multivariable analysis was not performed because >15% of data were missing for each variable, precluding comparison of our findings to the validated multiparametric prediction model published by Jansen et al.2
DIPG biology has been intensely studied since discovery of first-in-human histone mutations in 2012.15 Our findings confirm the independent association of H3.1 K27M and H3.3 K27M with long- and short-term survival, respectively.3,15 Median OS did not significantly differ between histone wild-type and mutant DIPGs; this contrasts with the report by Khuong-Quang et al15 of longer median OS (4.59 years) for patients with histone wild-type tumors.
In univariable analysis, WHO grade did not differ between LTSs and STSs (Table 1), but on Kaplan-Meier analysis, WHO grade 2 was associated with longer survival (Fig 3D). In the most recent WHO classification of CNS tumors,33 K27M-mutant midline gliomas are classified as WHO grade 4 regardless of histology, making this point less relevant. Tumors classified as primitive neuroectodermal tumors (now called embryonal tumor not otherwise specified) may represent true embryonal mimics of DIPG or result from sampling error in the context of intratumoral heterogeneity. Embryonal pontine tumors often demonstrate sharp margination and eccentric location, whereas others have radiologic characteristics indistinguishable from DIPG,34 like those excluded from our study (Appendix Table A2, online only).
A limitation of this study is use of disease-specific registry data, which are susceptible to enrollment bias on the part of participating institutions (which tend to be large academic centers) and patients or families who self-refer. Variation in standards of care between countries and institutions may have also influenced findings. Anonymity of registry data makes some overlap of registry patients with those previously reported possible, biasing our findings toward similarity with published literature because they are not completely independent cohorts. The primary strength of this study is mandated central review of diagnostic imaging with cross-validation by highly experienced pediatric neuroradiologists and use of standardized case report forms. To our knowledge, this study represents the largest, most comprehensively annotated cohort of radiographically confirmed DIPGs reported, offering the most accurate rates of long- and very long–term survival for this rare tumor. Identification of robust survival-associated factors in this study is vital for development of prognostic subgroups and emphasizes patient subsets from whom the most could be learned from analyzing pretreatment biopsy tissue. Understanding biologic differences that confer survival advantage in DIPG paevs the road toward development of subgroup-specific therapies that, when implemented in the context of clinical trials, may improve outcomes for this devastating disease.
Author Contributions
Conception and design: Lindsey M. Hoffman, Sophie E.M. Veldhuijzen van Zanten, Joshua Baugh, Adam Lane, James Leach, Dannis G. van Vuurden, Christof M. Kramm, Maryam Fouladi
Collection and assembly of data: Lindsey M. Hoffman, Sophie E.M. Veldhuijzen van Zanten, Niclas Colditz, Joshua Baugh, Brooklyn Chaney, Marion Hoffmann, Christine Fuller, Lili Miles, Cynthia Hawkins, Ute Bartels, Eric Bouffet, Stewart Goldman, Sarah Leary, Nicholas K. Foreman, Roger Packer, Katherine E. Warren, Alberto Broniscer, Mark W. Kieran, Jane Minturn, Melanie Comito, Emmett Broxson, Chie-Schin Shih, Soumen Khatua, Murali Chintagumpala, Anne Sophie Carret, Nancy Yanez Escorza, Timothy Hassall, David S. Ziegler, Nicholas Gottardo, Hetal Dholaria, Renee Doughman, Martin Benesch, Rachid Drissi, Javad Nazarian, Nathalie Boddaert, Pascale Varlet, G´eraldine Giraud, David Castel, Stephanie Puget, Chris Jones, Esther Hulleman, Piergiorgio Modena, Marzia Giagnacovo, Manila Antonelli, Torsten Pietsch, Gerrit H. Gielen, David T.W. Jones, Dominik Sturm, Stefan M. Pfister, Nicolas U. Gerber, Michael A. Grotzer, Elke Pfaff, Andr´e O. von Bueren, Darren Hargrave, Guirish A. Solanki, Filip Jadrijevic Cvrlje, Gertjan J.L. Kaspers, Jacques Grill, Simon Bailey, Veronica Biassoni, Maura Massimino, Rapha¨el Calmon, Brigitte Bison, Monika Warmuth-Metz, James Leach, Blaise Jones, Dannis G. van Vuurden, Christof M. Kramm, Maryam Fouladi
Data analysis and interpretation: Lindsey M. Hoffman, Sophie E.M. Veldhuijzen van Zanten, Joshua Baugh, Adam Lane, Christine Fuller, Lili Miles, Cynthia Hawkins, Nada Jabado, Esther Hulleman, Darren Hargrave, William P. Vandertop, Esther Sanchez, Brigitte Bison, Monika Warmuth- Metz, James Leach, Blaise Jones, Dannis G. van Vuurden, Christof M. Kramm, Maryam Fouladi
Manuscript writing: All authors.
Final approval of manuscript: All authors.
Accountable for all aspects of the work: All authors.
Authors' Disclosures of Potential Conflicts of Interest
Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Lindsey M. Hoffman No relationship to disclose
Sophie E.M. Veldhuijzen van Zanten No relationship to disclose
Niclas Colditz No relationship to disclose
Joshua Baugh No relationship to disclose
Brooklyn Chaney No relationship to disclose
Marion Hoffmann No relationship to disclose
Adam Lane No relationship to disclose
Christine Fuller No relationship to disclose
Lili Miles Honoraria: Aviana
Cynthia Hawkins Research Funding: Bayer
Patents, Royalties, Other Intellectual Property: RDLP 1001:Methods and application of low-grade glioma-specific genetic panel and analysis for clinical diagnostic and prognostic use; operationalized and implemented ependymoma CpG methylator phenotype (CIMP) subgrouping test in clinical setting; operationalized and implemented medulloblastoma subgroup test in clinical setting; GDF-15 as a prognostic biomarker for NK cell therapy; methods and application of sarcoma-specific genetic panel analysis for clinical diagnostic and prognostic use
Ute Bartels No relationship to disclose
Eric Bouffet Research Funding: Bristol-Myers Squibb (Inst), Roche (Inst)
Stewart Goldman Consulting or Advisory Role: Novartis
Travel, Accommodations, Expenses: Novartis
Sarah Leary No relationship to disclose
Nicholas K. Foreman No relationship to disclose
Roger Packer No relationship to disclose
Katherine E. Warren No relationship to disclose
Alberto Broniscer No relationship to disclose
Mark W. Kieran Consulting or Advisory Role: Novartis, Bayer, Boehringer Ingelheim,
Sigma Tau, Merck, Bristol-Myers Squibb, Takeda
Speakers’ Bureau: Merck Sharp & Dohme
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Jane Minturn No relationship to disclose
Melanie Comito No relationship to disclose
Emmett Broxson No relationship to disclose
Chie-Schin Shih Travel, Accommodations, Expenses: Exelixis
Soumen Khatua No relationship to disclose
Murali Chintagumpala No relationship to disclose
Anne Sophie Carret Employment: Seattle Genetics
Nancy Yanez Escorza No relationship to disclose
Timothy Hassall No relationship to disclose
David S. Ziegler No relationship to disclose
Nicholas Gottardo No relationship to disclose
Hetal Dholaria No relationship to disclose
Renee Doughman No relationship to disclose
Martin Benesch No relationship to disclose
Rachid Drissi Research Funding: PTC Therapeutics
Javad Nazarian No relationship to disclose
Nada Jabado No relationship to disclose
Nathalie Boddaert No relationship to disclose
Pascale Varlet Research Funding: Novartis (Inst), Boehringer Ingelheim (Inst), Roche (Inst)
G´eraldine Giraud No relationship to disclose
David Castel No relationship to disclose
Stephanie Puget No relationship to disclose
Chris Jones Consulting or Advisory Role: Roche
Research Funding: Roche
Travel, Accommodations, Expenses: Roche
Esther Hulleman No relationship to disclose
Piergiorgio Modena No relationship to disclose
Marzia Giagnacovo No relationship to disclose
Manila Antonelli No relationship to disclose
Torsten Pietsch Honoraria: Chugai Pharma
Travel, Accommodations, Expenses: Chugai Pharma
Gerrit H. Gielen No relationship to disclose
David T.W. Jones Patents, Royalties, Other Intellectual Property: Patent: Mutations of
histone proteins associated with proliferative disorders; patent pending:
DNA methylation-based method for classifying tumor species
Dominik Sturm No relationship to disclose
Stefan M. Pfister No relationship to disclose
Nicolas U. Gerber No relationship to disclose
Michael A. Grotzer No relationship to disclose
Elke Pfaff No relationship to disclose
Andr´e O. von Bueren No relationship to disclose
Darren Hargrave Consulting or Advisory Role: AstraZeneca, Genentech, Novartis, Bayer, Boehringer Ingelheim
Research Funding: AstraZeneca
Expert Testimony: AstraZeneca
Travel, Accommodations, Expenses: Boehringer Ingelheim, Novartis, Genentech
Other Relationship: Celgene, Novartis, Bristol-Myers Squibb, Epizyme, AbbVie
Guirish A. Solanki
Honoraria: Biomarin
Consulting or Advisory Role: Biomarin
Travel, Accommodations, Expenses: Biomarin
Filip Jadrijevic Cvrlje No relationship to disclose
Gertjan J.L. Kaspers No relationship to disclose
William P. Vandertop No relationship to disclose
Jacques Grill Research Funding: Roche, Novartis, Bristol-Myers Squibb
Simon Bailey No relationship to disclose
Veronica Biassoni No relationship to disclose
Maura Massimino Consulting or Advisory Role: Genentech
Rapha¨el Calmon No relationship to disclose
Esther Sanchez No relationship to disclose
Brigitte Bison No relationship to disclose
Monika Warmuth-Metz No relationship to disclose
James Leach No relationship to disclose
Blaise Jones No relationship to disclose
Dannis G. van Vuurden No relationship to disclose
Christof M. Kramm No relationship to disclose
Maryam Fouladi No relationship to disclose
Acknolwedgement
We thank the children and families who have suffered from diffuse intrinsic pontine glioma (DIPG) for their invaluable contribution to this research. Special thanks also to KPMG for the pro bono support of this project and for validation of design and security of the European Society for Pediatric Oncology (SIOPE) DIPG Registry and Imaging Repository.
Appendix
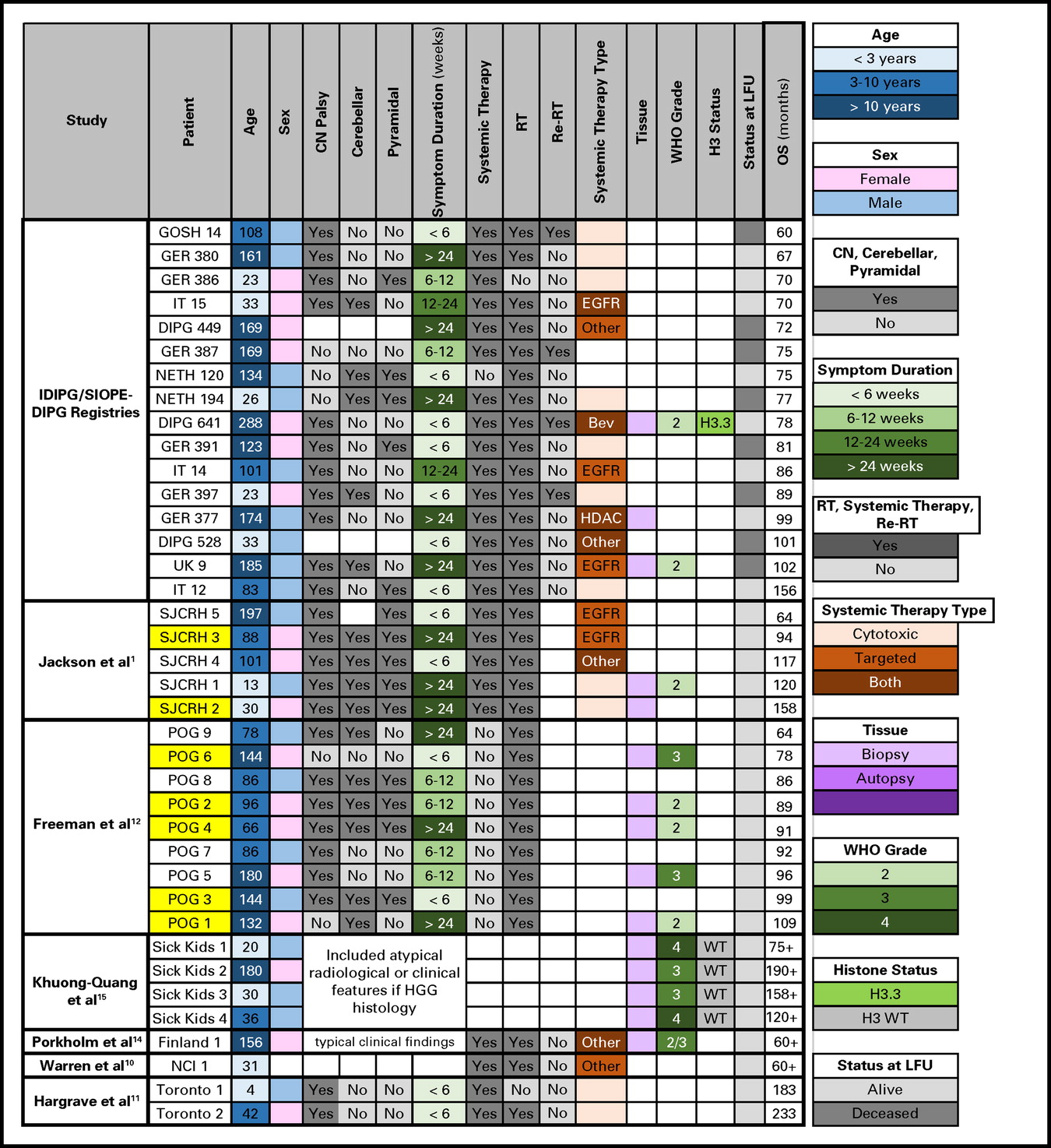
Very long–term survivors of diffuse intrinsic pontine glioma in the current study compared with those described in the literature. Yellow highlight indicates atypical radiologic features that would have been excluded in the current study. Bev, bevacizumab; CN, cranial nerve; DIPG, diffuse intrinsic pontine glioma; EGFR, epidermal growth factor; GER, Germany, Switzerland, Austria; GOSH, Great Ormond Street Hospital; HDAC, histone deacetylase inhibitor; HGG, high-grade glioma; IDIPGR, International Diffuse Intrinsic Pontine Glioma Registry; IT, Italy; LFU, last follow-up; NCI, National Cancer Institute; NETH, the Netherlands; OS, overall survival; POG, Pediatric Oncology Group; Re-RT, reirradiation; RT, radiation therapy; SIOPE, European Society for Pediatric Oncology; SJCRH, St Jude Children's Research Hospital; UK, United Kingdom; WT, wild type.
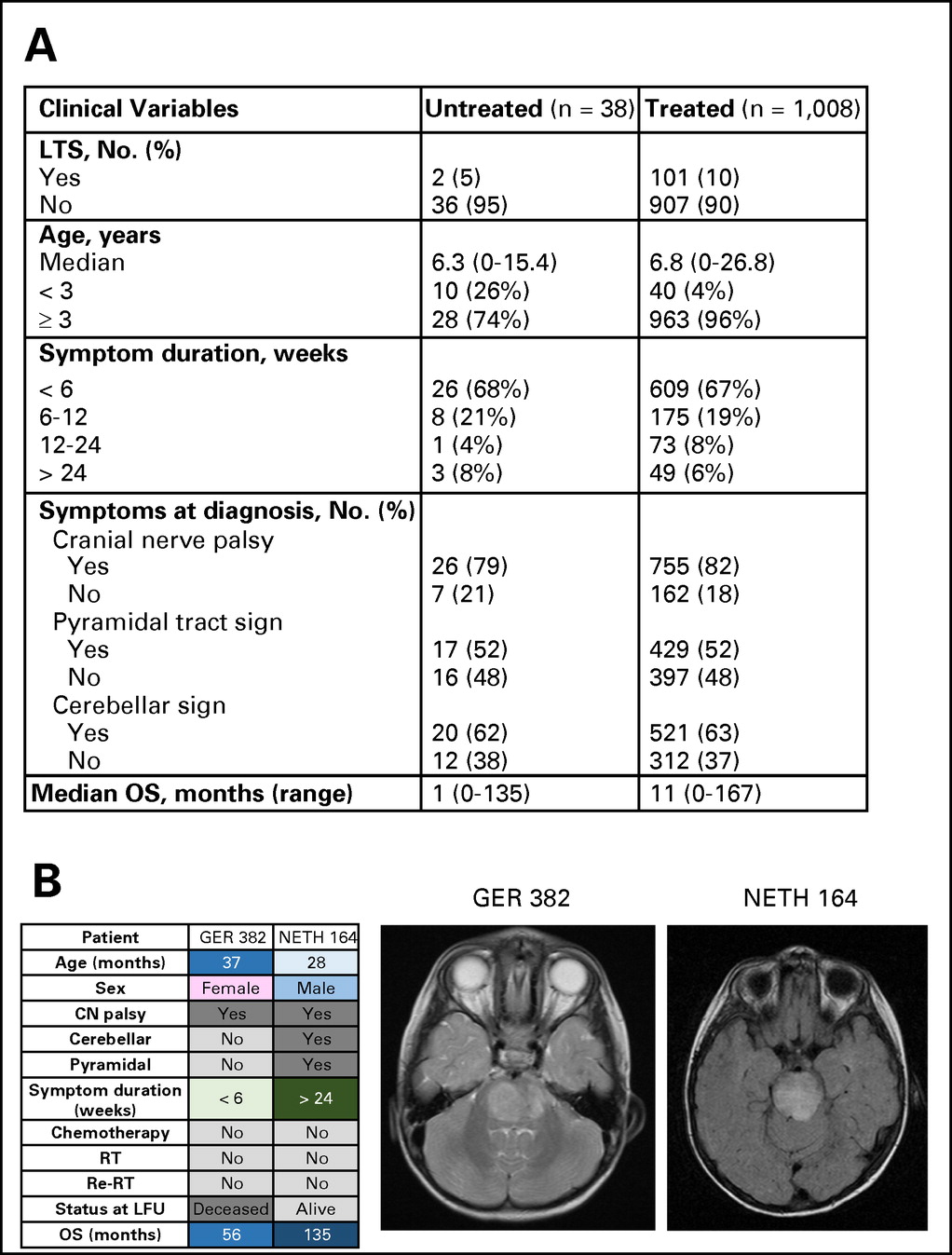
(A) Comparison of characteristics of patients who received therapy or did not receive therapy at diagnosis. (B) Magnetic resonance images and clinical characteristics of two long-term survivors (LTSs) of diffuse intrinsic pontine glioma who did not receive therapy. CN, cranial nerve; GER, Germany, Switzerland, Austria; LFU, last follow-up; NETH, the Netherlands; OS, overall survival; Re-RT, reirradiation; RT, radiation therapy.
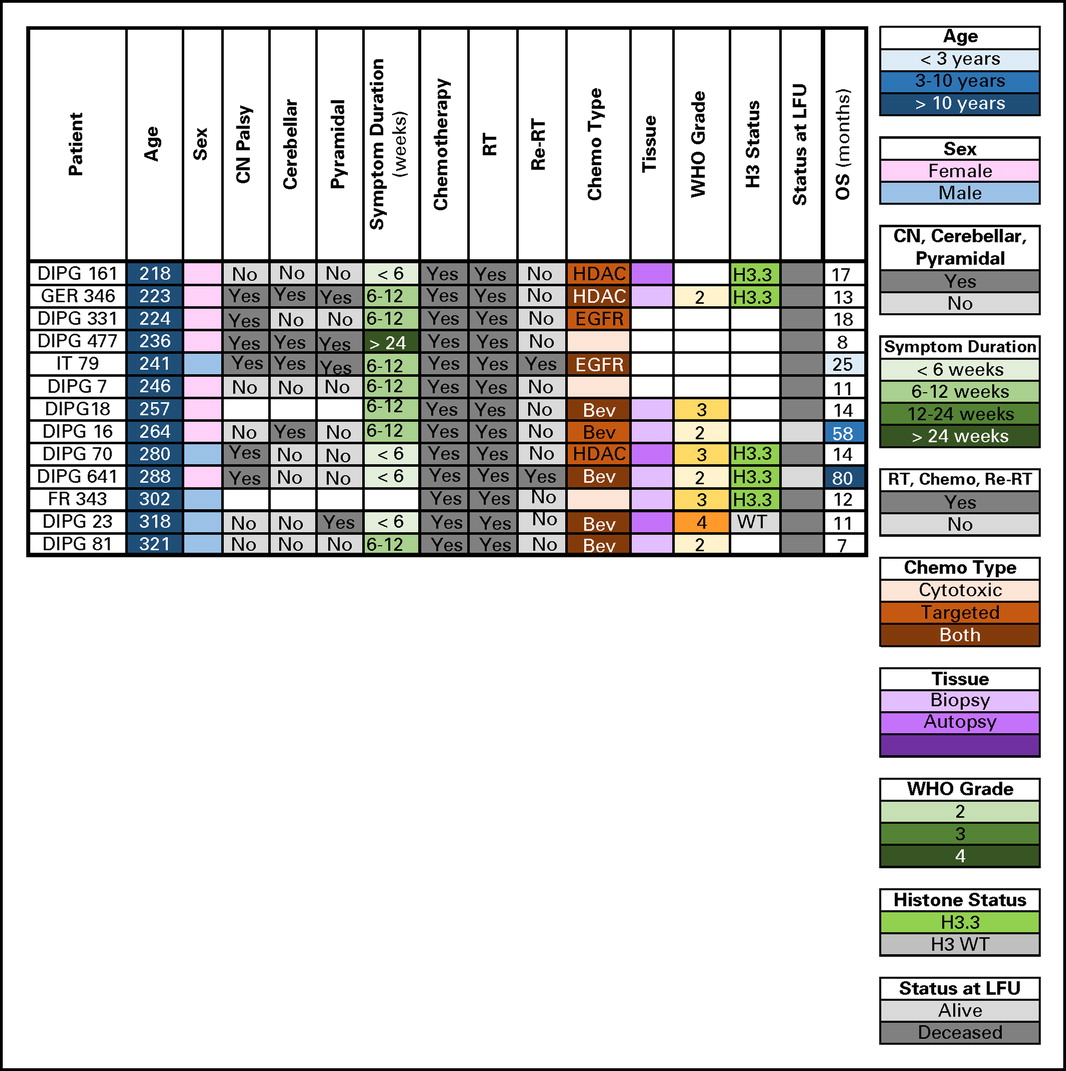
Clinical, radiologic, and molecular characteristics of patients with diffuse intrinsic pontine glioma age > 18 years. Bev, bevacizumab; CN, cranial nerve; DIPG, International DIPG Registry; EGFR, epidermal growth factor; FR, France; GER, Germany, Switzerland, Austria; HDAC, histone deacetylase inhibitor; IT, Italy; LFU, last follow-up; OS, overall survival; Re-RT, reirradiation; RT, radiation therapy; WT, wild type.
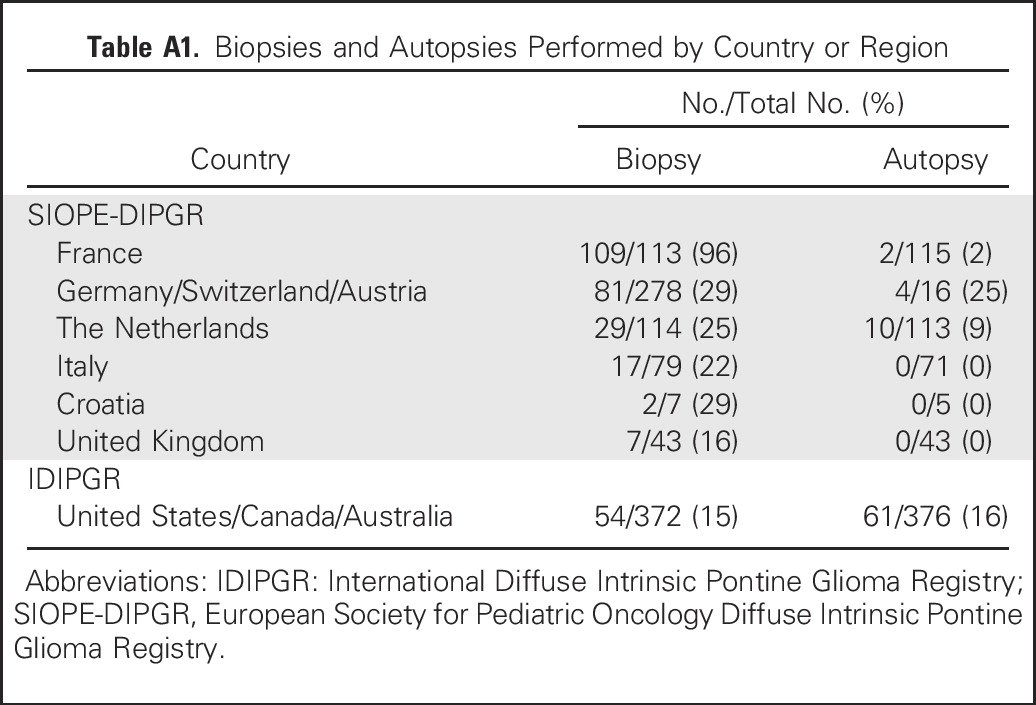
Biopsies and Autopsies Performed by Country or Region
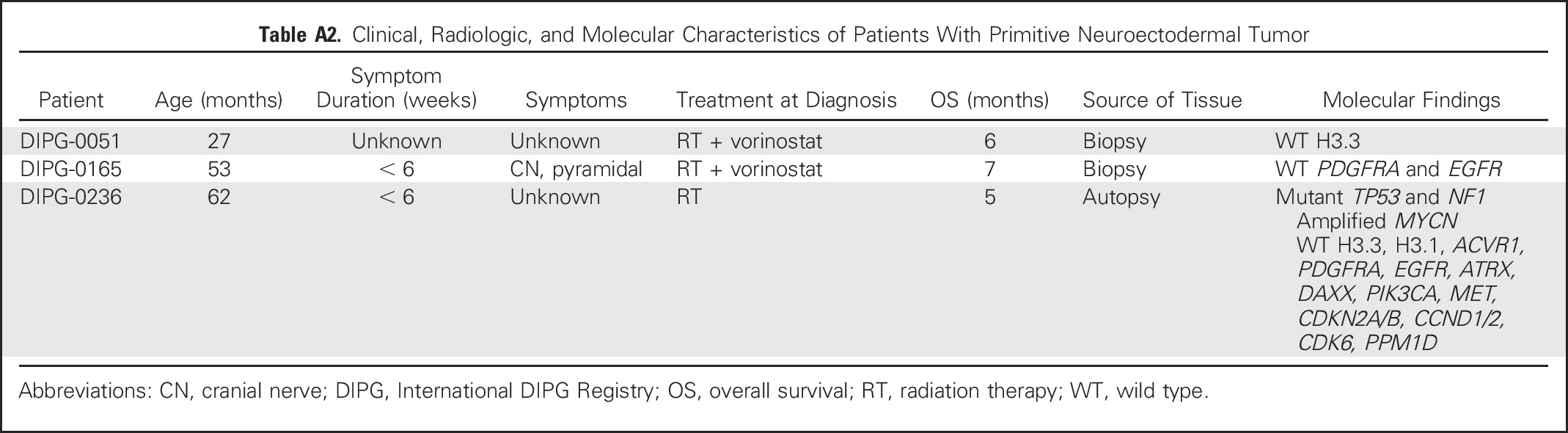
Clinical, Radiologic, and Molecular Characteristics of Patients With Primitive Neuroectodermal Tumor
References
- Jackson S, Patay Z, Howarth R, et al: Clinicoradiologic characteristics of long-term survivors of diffuse intrinsic pontine glioma. J Neurooncol 114:339-344, 2013
- Jansen MH, Veldhuijzen van Zanten SE, Sanchez Aliaga E, et al: Survival prediction model of children with diffuse intrinsic pontine glioma based on clinical and radiological criteria. Neuro Oncol 17:160-166, 2015
- Castel D, Philippe C, Calmon R, et al: Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130:815-827, 2015
- Gokce-Samar Z, Beuriat PA, Faure-Conter C, et al: Pre-radiation chemotherapy improves survival in pediatric diffuse intrinsic pontine gliomas. Childs Nerv Syst 32:1415-1423, 2016
- Wagner S, Warmuth-Metz M, Emser A, et al: Treatment options in childhood pontine gliomas. J Neurooncol 79:281-287, 2006
- Baugh J, Bartels U, Leach J, et al: The international diffuse intrinsic pontine glioma registry: An infrastructure to accelerate collaborative research for an orphan disease. J Neurooncol 132:323-331, 2017
- Veldhuijzen van Zanten SE, Baugh J, Chaney B, et al: Development of the SIOPE DIPG network, registry and imaging repository: A collaborative effort to optimize research into a rare and lethal disease. J Neurooncol 132:255-266, 2017
- Louis DN, Ohgaki H, Wiestler OD, et al: The 2007WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97-109, 2007
- Hassan H, Pinches A, Picton SV, et al: Survival rates and prognostic predictors of high grade brain stem gliomas in childhood: A systematic review and meta-analysis. J Neurooncol 135:13-20, 2017
- Warren K, Bent R, Wolters PL, et al: A phase 2 study of pegylated interferon a-2b (PEG-Intron) in children with diffuse intrinsic pontine glioma. Cancer 118:3607-3613, 2012
- Hargrave D, Chuang N, Bouffet E: Conventional MRI cannot predict survival in childhood diffuse intrinsic pontine glioma. J Neurooncol 86:313-319, 2008
- Freeman CR, Bourgouin PM, Sanford RA, et al: Long term survivors of childhood brain stem gliomas treated with hyperfractionated radiotherapy: Clinical characteristics and treatment related toxicities. Cancer 77:555-562, 1996
- Allen J, Siffert J, Donahue B, et al: A phase I/II study of carboplatin combined with hyperfractionated radiotherapy for brainstem gliomas. Cancer 86:1064-1069, 1999
- Porkholm M, Valanne L, Lo¨nnqvist T, et al: Radiation therapy and concurrent topotecan followed by maintenance triple anti-angiogenic therapy with thalidomide, etoposide, and celecoxib for pediatric diffuse intrinsic pontine glioma. Pediatric Blood Cancer 61:1603-1609, 2014
- Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, et al: K27Mmutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439-447, 2012
- Veldhuijzen van Zanten SEM, Jansen MHA, Sanchez Aliaga E, et al: A twenty-year review of diagnosing and treating children with diffuse intrinsic pontine glioma in the Netherlands. Expert Rev Anticancer Ther 15:157-164, 2015
- Broniscer A, Laningham FH, Sanders RP, et al: Young age may predict a better outcome for children with diffuse pontine glioma. Cancer 113:566-572, 2008
- Wu G, Diaz AK, Paugh BS, et al: The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstemhigh-grade glioma. Nat Genet 46:444-450, 2014
- Bailey S, Howman A, Wheatley K, et al: Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy: Results of a United Kingdom phase II trial (CNS 2007 04). Eur J Cancer 49:3856-3862, 2013
- Fisher PG, Breiter SN, Carson BS, et al: A clinicopathologic reappraisal of brain stem tumor classification: Identification of pilocystic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer 89:1569-1576, 2000
- Hargrave D, Bartels U, Bouffet E: Diffuse brainstem glioma in children: Critical review of clinical trials. Lancet Oncol 7:241-248, 2006
- JansenMHA, van Vuurden DG, VandertopWP, et al: Diffuse intrinsic pontine gliomas: A systematic update on clinical trials and biology. Cancer Treat Rev 38:27-35, 2012
- Pollack IF, Stewart CF, KocakM, et al: A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: A report from the Pediatric Brain Tumor Consortium. Neuro Oncol 13:290-297, 2011
- Geoerger B, Hargrave D, Thomas F, et al: Innovative Therapies for Children with Cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro Oncol 13: 109-118, 2011
- Bartels U, Wolff J, Gore L, et al: Phase 2 study of safety and efficacy of nimotuzumab in pediatric patients with progressive diffuse intrinsic pontine glioma. Neuro Oncol 16:1554-1559, 2014
- Hummel TR, Salloum R, Drissi R, et al: A pilot study of bevacizumab-based therapy in patients with newly diagnosed high-grade gliomas and diffuse intrinsic pontine gliomas. J Neurooncol 127:53-61, 2016
- Gururangan S, Fangusaro J, Poussaint TY, et al: Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas: A Pediatric Brain Tumor Consortium study. Neuro Oncol 16:310-317, 2014
- Broniscer A, Baker JN, Tagen M, et al: Phase I study of vandetanib during and after radiotherapy in children with diffuse intrinsic pontine glioma. J Clin Oncol 28:4762-4768, 2010
- Janssens GO, Gandola L, Bolle S, et al: Survival benefit for patients with diffuse intrinsic pontine glioma (DIPG) undergoing re-irradiation at first progression: A matched-cohort analysis on behalf of the SIOP-E-HGG/DIPG working group. Eur J Cancer 73: 38-47, 2017
- Morales La Madrid A, Santa-Mar´ıa V, Cruz Martinez O, et al: Second re-irradiation for DIPG jco.org © 2018 by American Society of Clinical Oncology 1971 Long-Term Survivors of Diffuse Intrinsic Pontine Glioma Downloaded from ascopubs.org by Children's Hospital on September 2, 2019 from 205.142.197.113 Copyright © 2019 American Society of Clinical Oncology. All rights reserved. progression, re-considering “old strategies” with new approaches. Childs Nerv Syst 33:849-852, 2017
- Barkovich AJ, Krischer J, Kun LE, et al: Brain stem gliomas: A classification system based on magnetic resonance imaging. Pediatr Neurosurg 16:73-83, 1990-1991
- Poussaint TY, Kocak M, Vajapeyam S, et al: MRI as a central component of clinical trials analysis in brainstem glioma: A report from the Pediatric Brain Tumor Consortium (PBTC). Neuro Oncol 13:417-427, 2011
- David N, Louis MD, Ohgaki H: WHO Classification of Tumours of the Central Nervous System. Geneva, Switzerland, World Health Organization, 2007
- Sufit A, Donson AM, Birks DK, et al: Diffuse intrinsic pontine tumors: A study of primitive neuroectodermal tumors versus the more common diffuse intrinsic pontine gliomas. J Neurosurg Pediatr 10:81-88, 2012

